Мопатхић - дијагноза озбиљног. У овом чланку, практичар говори о врстама ове многостране болести.
Садржај
 «Ми смо три браће (31 година стара, 29 и 27 година) - са адолесценцијом патим од једне болести - прогресивна мишића дистрофија. Сва три инвалида. Можда ће стручњаци бити враћени на нашој несрећи и помоћи.»
«Ми смо три браће (31 година стара, 29 и 27 година) - са адолесценцијом патим од једне болести - прогресивна мишића дистрофија. Сва три инвалида. Можда ће стручњаци бити враћени на нашој несрећи и помоћи.»
«У детету (10 година), прогресивна мишићна дистрофија Дукеда. Лекари немоћни. Бићу захвалан на било којим рецептима, саветима.»
«Унук (3 године) изненада је почео да негира ноге и током времена се погоршава. Љекари су ставили различите дијагнозе и не могу ништа да ураде. Помозите добрим саветима.»
То су одломци из писма људи који су се сударили са тако озбиљном болешћу као миопатија. Шта је миопатија? Покушајмо да класификујемо овај мулти-графикон.
Миопатија и њене врсте
Моопатхи представља групу неуромускуларних болести које се манифестују умора, слабост мишића, смањење мишићног тона, мишићног атрофије. Моопатхи, у зависности од узрочности, подељен је у прогресивну наследну мишићну дистрофију, ендокрине миопатије (болести унутрашњих секреција жлезда) и метаболичке миопатије (метаболички поремећаји).
Разговарајте о прогресивном наследној дистрофији мишића. Ову врсту миопатије карактерише мишићна атрофија због уништавања мишићних ћелија због недостатка посебног протеина, што јача структуру мишићних влакана. Овај протеин се производи под контролом посебног ћелијског гена, који се налази на 6. хуманом хромозому, а током квара овог гена долази постепено уништавање шкољки мишићних ћелија, праћено бацањем мишићних влакана.
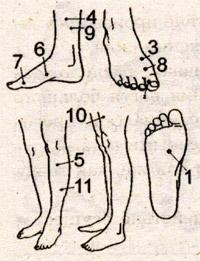
Овај неисправан ген је наслеђен ако је било брак између рођака. Промене гена у 30% случајева настаје као резултат мутације, односно у овим случајевима брак између рођака - нити. Болест је наслеђена са 50% вероватноће, ако је један од родитеља детета болестан. Повезано је са женским сексуалним хромозом и преноси се по правилу синова, иако жене сами можда нису повређене. Атрофија Мишићи ручица рамена, леђа, карлични појас и ноге.
У зависности од локализације болести, старости, тежина болести издваја различите облике мишићне дистрофије. Дакле, облик омладинске ротера дешава се у доби од 10-20 година, када се атрофија мишића раменог појаса и руку појављује неприметно, а затим - карлични појас и ноге. Док је ходао пацијента, трбух и уврнут задњи део груди. Да се истакнете са положаја, пацијент се окреће са своје стране и, наслонивши руке на бокове, постепено подиже своје тело. Болест полако напредује.
Дечији облик мишићне дистрофије Дудеда почиње у доби од 3-5 година атрофијом мишића карлице, боковима са истодобним задебљањем Осцрацланал мишића ноге (лажно задебљање). Постепено атрофија мишића рамена каишева и руку. Деца у почетку узнемирила ход, а затим постављају потешкоће у покрету. Многи имају откуцаја срца због повећања величине срца. Прогресија болести или њен малигни проток због раног имобилизације удова доводи до тужног исхода. Они су болесни, углавном дечаци (1 за 3000 рођених). Да би били тачнији, мушкарци и жене су такође болесни. Само Досенова болест се манифестује у момцима. Дјевојке су носилаци овог гена.
Али дешава се и бенигне за мишићну дистрофију (Бецкер миодастрофију), када се болест манифестује полако, посебно у дјеци са мало духовима. Дуги низ година задржавају задовољавајуће физичко стање и само приступање различитих акутних болести и повреда доводи их на имобилизацију, исцрпљивање са лошим исходом.
Облик лица-лица у мионитрофију, зван Ландузи-Дезхард, који може бити стари 6 до 52 године (чешће у 10-15 година) и карактерише је пораз мишића лица са постепеним накнадним атрофијом мишићи рамена, торзо и удова. Рани знакови болести су слабо затворени и затворени капци, потпуно затворени усне, што ствара нејасан говор и немогућност надувавања образа. Болест се полако наставља. Дуго се пацијент може преместити и одржавати способност рада, а после 15-25 година, мишићи карличног појаса ногу постепено су атрофизирани, што отежава премештање.
Такође је додељена група секундарне прогресивне мишићне дистрофије која се појављује у вези са оштећењем нераваца: неурол, кичмена миодастрофија која се ипак назива и даље амиотрофија.
Амиотрофија Схарк-Марие Амиотрофија, коју карактерише постепена атрофија малих мишића заустављања, тада су атрофија мишићи ногу и доњи део кукова, а мишићи средње и горње делове бокова не промена и бедро је облик боце са вратом, нагнут. Мишићи руку и подлактице су потом постепено атрофија. Мишићи торза, каишева и лица. Болест се јавља у доби од 18-25 година, полако напредује и стабилизује.
Конгенитална кидна мишићна атрофија Кугелберга-Валлангер карактерише постепена атрофија мишића руку, ногу, ретардације менталног и физичког развоја, деформација кичме. Болест се манифестује у доби од 8-10 година и полако напредује.
Прогресивна аитропонија Аран-Дужена почиње у доби од 25-50 година и манифестује атрофију мишића четкице. Тада су остали руке постепено атрофизирани, а затим стопала тела, у т.Ц. интеркостални мишићи, који изазива респираторне поремећаје из које дође смрт.
Конгенитална амиотонија (смањење мишићног тона) Оппенхеим карактерише слабост мишића због њихове неразвијености и њихова мишићна дистрофија је секундарна. У новорођенчету, то не напредује, али улазак респираторних инфекција може проузроковати упалу и смрт долази у првој години живота. Са годинама, функција мотора мишића се побољшава.
Лечење мишићне дистрофије је усмерен на успоравање дистрофичних (уништавајућих) процеса у мишићима, па чак и њихово престанак. Међутим, радикално лечење још увек није пронађено. Иако је нада на генинској терапији, која почиње да полако спроводи у медицинску праксу.